




What’s GAMP?
The Good Automated Manufacturing Practice (GAMP)
Forum was founded in 1991 by pharmaceutical industry professionals in
the United Kingdom to address the industry’s need to improve
comprehension and evolving expectations of regulatory agencies in
Europe. The organization also sought to promote understanding of how
computer systems validation should be conducted in the pharmaceutical
industry.
In 1994, GAMP partnered with the International Society for Pharmaceutical Engineering (ISPE) to publish the first GAMP guidelines. GAMP quickly became influential throughout Europe as the quality of its work was recognized internationally. Over time, GAMP has become the acknowledged expert body for addressing issues of computer system validation.
GAMP's guidance approach defines a set of industry best practices to enable compliance to all current regulatory expectations. More than simply a strict compliance standard, GAMP is a guideline for life sciences companies to use for their own quality procedures. As a result, it can be tailored to a number of computer system types.
Computer system validation following GAMP guidelines requires users and suppliers to work in concert so that responsibilities regarding the validation process are understood. For users, GAMP provides a documented assurance that a system is appropriate for the intended use before it goes “live.” Suppliers can use GAMP to test for avoidable defects in the supplied system to ensure quality product leaves the facility.
The GAMP framework addresses how systems are validated and documented, in other words “how one will validate and document the system.” Companies do not need to follow the same set of procedures and processes of a GAMP framework to achieve validation and qualification levels that satisfy inspectors. Instead, GAMP examines the systems development lifecycle (SDLC) – a conceptual model that lays out the deliverable documents required by GAMP – of an automated system to identify issues of validation, compliance and documentation.
In essence, GAMP asks:
* Do you know what you want to do?
* Have you broadly defined the function requirements?
* How will you do it?
Identifying the “how” is essential to the design and testing phases of validation. Once the design is tested, and if it works as intended, then you have satisfied not only the function requirements, but the overall requirements for system use. A regulatory body expects to see documentation of the process.
In 1994, GAMP partnered with the International Society for Pharmaceutical Engineering (ISPE) to publish the first GAMP guidelines. GAMP quickly became influential throughout Europe as the quality of its work was recognized internationally. Over time, GAMP has become the acknowledged expert body for addressing issues of computer system validation.
GAMP's guidance approach defines a set of industry best practices to enable compliance to all current regulatory expectations. More than simply a strict compliance standard, GAMP is a guideline for life sciences companies to use for their own quality procedures. As a result, it can be tailored to a number of computer system types.
Computer system validation following GAMP guidelines requires users and suppliers to work in concert so that responsibilities regarding the validation process are understood. For users, GAMP provides a documented assurance that a system is appropriate for the intended use before it goes “live.” Suppliers can use GAMP to test for avoidable defects in the supplied system to ensure quality product leaves the facility.
The GAMP framework addresses how systems are validated and documented, in other words “how one will validate and document the system.” Companies do not need to follow the same set of procedures and processes of a GAMP framework to achieve validation and qualification levels that satisfy inspectors. Instead, GAMP examines the systems development lifecycle (SDLC) – a conceptual model that lays out the deliverable documents required by GAMP – of an automated system to identify issues of validation, compliance and documentation.
In essence, GAMP asks:
* Do you know what you want to do?
* Have you broadly defined the function requirements?
* How will you do it?
Identifying the “how” is essential to the design and testing phases of validation. Once the design is tested, and if it works as intended, then you have satisfied not only the function requirements, but the overall requirements for system use. A regulatory body expects to see documentation of the process.
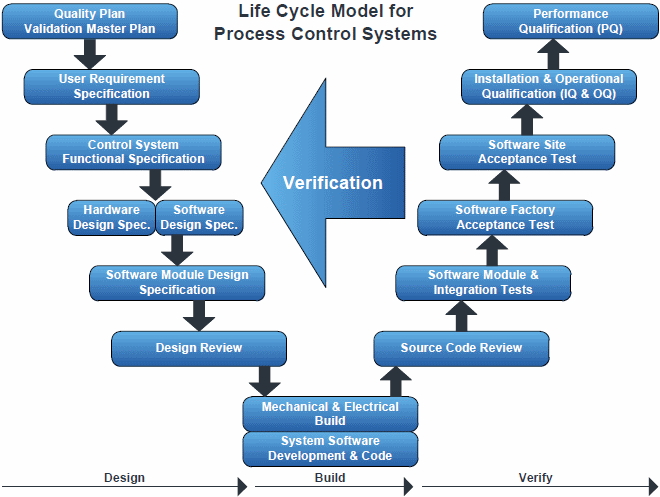
‘V’ for Validation
GAMP
recommends an SDLC called the V-model (see graphic) because it is a
commonly used design, but there are others that can be followed. The
V-model shows how the three main qualification activities (installation,
operation and performance) are linked back to the design process.
These main steps correspond to deliverables within a computerized validation framework. The left side of the V represents the specification stream – user requirements, functional specifications, hardware and software design, and module specifications. The right side of the V represents the system testing stream against the specifications. The bottom of the V indicates the code modules.
These main steps correspond to deliverables within a computerized validation framework. The left side of the V represents the specification stream – user requirements, functional specifications, hardware and software design, and module specifications. The right side of the V represents the system testing stream against the specifications. The bottom of the V indicates the code modules.
Specification Stream
With the
V-model, the document that initiates the validation process is the user
requirement specification (URS). The URS describes the equipment or
system as it is intended to function, and it is typically written by the
system user. The original version should contain the essential
requirements and the desirable requirements. As part of the validation
process, the organization checks the software system before launch.
Clear documentation of a properly functioning system is typically found
in the URS to detail what the system should do and what it could do.
Next, the URS is matched with the functional and design specifications, which often come from the system or software developer. The functional specifications describe the functions of the system and how it was built. In the V-model, the functional specifications correspond to the operational qualifications, as each of the parameters should be tested. A gap analysis is performed to identify areas where an internal requirement isn’t met. This allows recognition of risks and outlines approaches to correct the shortcomings. The design specifications define the production of the hardware, software and instrumentation and how the software meets the requirements of the functional specifications for proper function.
Next, the URS is matched with the functional and design specifications, which often come from the system or software developer. The functional specifications describe the functions of the system and how it was built. In the V-model, the functional specifications correspond to the operational qualifications, as each of the parameters should be tested. A gap analysis is performed to identify areas where an internal requirement isn’t met. This allows recognition of risks and outlines approaches to correct the shortcomings. The design specifications define the production of the hardware, software and instrumentation and how the software meets the requirements of the functional specifications for proper function.
Testing Stream
Validation is applied to several
aspects of a pharmaceutical manufacturing system. The objective is to
produce “documented evidence, which provides a high degree of assurance
that all parts of a system will consistently work correctly when brought
on-line. Validation includes three core elements:
* Installation qualification (IQ) – confirms complete documentation, which includes checking purchase orders, proper hardware installation, and software verification according to the manufacturer’s specifications; both user and supplier share primary testing responsibility.
* Operational qualification (OQ) – confirms the system operations by testing the design requirements that are traced back to the function specifications, including software and hardware functions under normal load, and under realistic stress conditions to assess whether equipment and systems are working correctly; both user and supplier share primary testing responsibility.
* Performance qualification (PQ) – confirms that a system is capable of performing or controlling the activities of the process, while operating in a specific environment – namely, a series of checks by the user against the original requirement specifications of the system; responsibility falls solely on the user.
Though there isn’t a singular method for achieving and maintaining traceability, regulatory agencies have an essential level of expectation. Despite the lack of a standard procedure, the selected process and method used by a system for traceability should be documented and understood. The core principles of traceability link system requirements, design specifications and testing documents with the processes and supporting documentation. In other words, traceability should demonstrate that by testing the documents, one is able to verify the system requirements a nd the design specifications.
The linkage among requirements, design and testing may be identified by the following relationships:
* Multiple requirements may be checked by a single design specification and confirmed by a single test;
* Multiple design specifications may be coupled to a single requirement; and
* Multiple tests may be necessary to verify one requirement or one design specification.
Traceability may be achieved through:
* A requirements traceability matrix;
* Automated software tools; and
* Embedded references directly within documents.
Organizations use GAMP guidelines to achieve traceability by checking whether a system is:
* Appropriate in its size, complexity, impact and risk;
* Documented and approved in the validation planning stage; and
* Integral to the overall project life cycle and for the support and maintenance of the system.
* Installation qualification (IQ) – confirms complete documentation, which includes checking purchase orders, proper hardware installation, and software verification according to the manufacturer’s specifications; both user and supplier share primary testing responsibility.
* Operational qualification (OQ) – confirms the system operations by testing the design requirements that are traced back to the function specifications, including software and hardware functions under normal load, and under realistic stress conditions to assess whether equipment and systems are working correctly; both user and supplier share primary testing responsibility.
* Performance qualification (PQ) – confirms that a system is capable of performing or controlling the activities of the process, while operating in a specific environment – namely, a series of checks by the user against the original requirement specifications of the system; responsibility falls solely on the user.
Though there isn’t a singular method for achieving and maintaining traceability, regulatory agencies have an essential level of expectation. Despite the lack of a standard procedure, the selected process and method used by a system for traceability should be documented and understood. The core principles of traceability link system requirements, design specifications and testing documents with the processes and supporting documentation. In other words, traceability should demonstrate that by testing the documents, one is able to verify the system requirements a nd the design specifications.
The linkage among requirements, design and testing may be identified by the following relationships:
* Multiple requirements may be checked by a single design specification and confirmed by a single test;
* Multiple design specifications may be coupled to a single requirement; and
* Multiple tests may be necessary to verify one requirement or one design specification.
Traceability may be achieved through:
* A requirements traceability matrix;
* Automated software tools; and
* Embedded references directly within documents.
Organizations use GAMP guidelines to achieve traceability by checking whether a system is:
* Appropriate in its size, complexity, impact and risk;
* Documented and approved in the validation planning stage; and
* Integral to the overall project life cycle and for the support and maintenance of the system.
Top Three Challenges
As
a voluntary program, GAMP offers both challenges and benefits. The top
three challenges in implementing GAMP are establishing procedural
control, handling management and change control, and finding an
acceptable standard among the existing variations.
Establishing procedural control is a challenge in using GAMP guidelines because new frameworks may be necessary to gauge the validity of systems. Most pharmaceutical companies have already established a baseline that adheres to standards and regulations that exist today, but they may not have a procedure to check the processes that are in place. This could cause resistance among software developers who may prefer not to work within the confines of specifications and procedures developed by others. Specifications and procedures developed by previous software developers may hinder ways to adjust computer systems, but varying interpretations of GAMP guidelines allow for multiple solutions.
Another hurdle is change control. In the development or modification of computer systems, companies with even the highest of standards can suffer setbacks along the SDLC. Sometimes minor tweaks by the software programmer, whether necessary or not, may cause breakdowns after validation changes have been implemented. Internal processes and procedures must be established to guard against these occurrences.
Whether utilizing another company’s specifications and procedures or your own, effective documentation management is fundamental for compliance. Any inaccuracies or missing information renders all other efforts moot. Moreover, implementing a formal document management application may be cost-prohibitive for some organizations. Some companies simply use what’s in the GAMP checklists to evaluate their systems. Today’s environment demands a thorough process to show validation.
Establishing procedural control is a challenge in using GAMP guidelines because new frameworks may be necessary to gauge the validity of systems. Most pharmaceutical companies have already established a baseline that adheres to standards and regulations that exist today, but they may not have a procedure to check the processes that are in place. This could cause resistance among software developers who may prefer not to work within the confines of specifications and procedures developed by others. Specifications and procedures developed by previous software developers may hinder ways to adjust computer systems, but varying interpretations of GAMP guidelines allow for multiple solutions.
Another hurdle is change control. In the development or modification of computer systems, companies with even the highest of standards can suffer setbacks along the SDLC. Sometimes minor tweaks by the software programmer, whether necessary or not, may cause breakdowns after validation changes have been implemented. Internal processes and procedures must be established to guard against these occurrences.
Whether utilizing another company’s specifications and procedures or your own, effective documentation management is fundamental for compliance. Any inaccuracies or missing information renders all other efforts moot. Moreover, implementing a formal document management application may be cost-prohibitive for some organizations. Some companies simply use what’s in the GAMP checklists to evaluate their systems. Today’s environment demands a thorough process to show validation.
Get GAMP
How
do companies become GAMP-aware when it comes to dealing with the
variability of process and procedures that exist in the industry? Some
manufacturers that operate plants in numerous locations have established
their own set of specifications and procedures to follow GAMP
guidelines, and may add and drop some criteria to dictate the level of
validation necessary to work with them. Suppliers reference GAMP because
they’re following another company’s pre-established procedures. The
customer can dictate changes to the supplier if they are necessary.
The ubiquitous pharmaceutical industry deals with not only domestic and international companies, but also a number of regulatory bodies as well. Inevitably, they’re facing some code of federal regulations along with GAMP, especially when a company wishes to export to the U.S., Europe and other parts of the world.
There are many companies that are capable of validating their systems to their specifications because they know they have to satisfy the FDA and have aligned their efforts accordingly. However, the FDA requirements are not prescriptive with step-by-step procedures, but are guidelines with an approximation of checks and balances. Some companies demonstrate validation by documenting the process to make a product consistent and repeatable to their own specifications. In some cases, companies simply follow what the customer wants. The lack of a rigid guideline should signal to companies that some give-and-take is necessary - whether satisfying customers or regulatory agencies.
The ubiquitous pharmaceutical industry deals with not only domestic and international companies, but also a number of regulatory bodies as well. Inevitably, they’re facing some code of federal regulations along with GAMP, especially when a company wishes to export to the U.S., Europe and other parts of the world.
There are many companies that are capable of validating their systems to their specifications because they know they have to satisfy the FDA and have aligned their efforts accordingly. However, the FDA requirements are not prescriptive with step-by-step procedures, but are guidelines with an approximation of checks and balances. Some companies demonstrate validation by documenting the process to make a product consistent and repeatable to their own specifications. In some cases, companies simply follow what the customer wants. The lack of a rigid guideline should signal to companies that some give-and-take is necessary - whether satisfying customers or regulatory agencies.
What Do I Need?
If a life
sciences company wishes to use GAMP guidelines to set up its validation
systems, some of the elements may already be in place. Certain aspects,
such as the maturity of the hardware or software, must be taken into
consideration to check whether these elements are “industry proven.” To
test the validity of elements in the system, the appropriate hardware,
infrastructure and network must be in place. When beginning the testing
environment, the test author should understand the testing environment
in terms of:
* Correct hardware (peripherals and interfaces);
* Software (validated tools, software configuration);
* Data units (inputs, outputs, quality and quantity of data);
* Procedures (especially for user acceptance testing); and
* People (training and experience), (GAMP Good Practice Guide, pg. 69).
Suppliers can offer highly scalable automation architectures, which can be applied to a stand-alone one-server/one-user application, or to multiple users interfacing with multiple servers. This allows companies the ability to improve flexibility, reduce downtime and improve productivity. For example, a database system that wasn’t 21 CFR Part 11-compliant would require the company to make adjustments to the computer system to become compliant. This means the automation infrastructure must drive regulatory compliance to ensure that products meet guidelines. Likewise, OEMs are now looking at ways to provide the pro forma operational qualifications for all features in their equipment, so companies can test each of the features. Likewise, automation suppliers offer technology enhancements, as well as parts, small systems, total systems and integrated systems to help streamline the qualification process and reduce validation costs.
Typically, the costs of validating a larger system often represent between 20-25% of the total cost of the system qualification. Reducing the cost adds value to the bottom line and enables a system to go on-line faster. It makes sense to have procedures and systems in place to make validation easier.
GAMP helps companies address current issues of operational/manufacturing challenges through standardizing data, monitoring systems and validating the system.
The benefits of utilizing the GAMP approach for both users and suppliers include:
* Improved understanding of the subject with the introduction of common terminology;
* Reduced cost and time to achieve compliant systems;
* Reduced time and resources for revalidation or regression testing and remediation;
* Reduced cost of qualification;
* Enhanced compliance with regulatory expectations; and
* Established responsibility for all involved parties.
Products are available to help companies avoid revalidating an entire system when a new version emerges. Software tools focused on the life sciences industry that support cost-effective, risk-based manufacturing approaches allow companies to see what testing has been done to examine the functions within the system.
When the FDA introduced its current Good Manufacturing Practices (cGMP) for the 21st century initiative, companies shifted their approach to validation. Formerly, they only had to heed a set of rules that accounted for every piece of equipment that was used. Now they can take a risk-based approach to validation by addressing patient safety, efficacy and quality in the product considerations. In essence, this enables the industry to place its investments where it makes the most sense. The onus ultimately falls on manufacturers to accept greater responsibility to validate their systems having the attendant benefits of cost and time to market savings.
GAMP helps provide a quality product from the manufacturer, and helps to limit the pharmaceutical industry’s culpability by ensuring proper steps were placed to deliver a quality product through validated systems. By incorporating input from the full spectrum of stakeholders, fine tuning and further development of the process is geared towards benefiting the life sciences industry and the general consumer market.
The tools exist for companies to take the steps needed to reap the benefits of validation. Clearly, if you aren’t taking the necessary steps to compete, then your competitors are assuredly doing what they can to gain a market advantage. Understanding and early adoption of GAMP can increase a company’s competitive position, especially with the introduction/implementation of new technologies. By staying aware of technological innovations, companies are able to increase efficiency, minimize risks and reduce costs.
* Correct hardware (peripherals and interfaces);
* Software (validated tools, software configuration);
* Data units (inputs, outputs, quality and quantity of data);
* Procedures (especially for user acceptance testing); and
* People (training and experience), (GAMP Good Practice Guide, pg. 69).
Suppliers can offer highly scalable automation architectures, which can be applied to a stand-alone one-server/one-user application, or to multiple users interfacing with multiple servers. This allows companies the ability to improve flexibility, reduce downtime and improve productivity. For example, a database system that wasn’t 21 CFR Part 11-compliant would require the company to make adjustments to the computer system to become compliant. This means the automation infrastructure must drive regulatory compliance to ensure that products meet guidelines. Likewise, OEMs are now looking at ways to provide the pro forma operational qualifications for all features in their equipment, so companies can test each of the features. Likewise, automation suppliers offer technology enhancements, as well as parts, small systems, total systems and integrated systems to help streamline the qualification process and reduce validation costs.
Typically, the costs of validating a larger system often represent between 20-25% of the total cost of the system qualification. Reducing the cost adds value to the bottom line and enables a system to go on-line faster. It makes sense to have procedures and systems in place to make validation easier.
GAMP helps companies address current issues of operational/manufacturing challenges through standardizing data, monitoring systems and validating the system.
The benefits of utilizing the GAMP approach for both users and suppliers include:
* Improved understanding of the subject with the introduction of common terminology;
* Reduced cost and time to achieve compliant systems;
* Reduced time and resources for revalidation or regression testing and remediation;
* Reduced cost of qualification;
* Enhanced compliance with regulatory expectations; and
* Established responsibility for all involved parties.
Products are available to help companies avoid revalidating an entire system when a new version emerges. Software tools focused on the life sciences industry that support cost-effective, risk-based manufacturing approaches allow companies to see what testing has been done to examine the functions within the system.
When the FDA introduced its current Good Manufacturing Practices (cGMP) for the 21st century initiative, companies shifted their approach to validation. Formerly, they only had to heed a set of rules that accounted for every piece of equipment that was used. Now they can take a risk-based approach to validation by addressing patient safety, efficacy and quality in the product considerations. In essence, this enables the industry to place its investments where it makes the most sense. The onus ultimately falls on manufacturers to accept greater responsibility to validate their systems having the attendant benefits of cost and time to market savings.
GAMP helps provide a quality product from the manufacturer, and helps to limit the pharmaceutical industry’s culpability by ensuring proper steps were placed to deliver a quality product through validated systems. By incorporating input from the full spectrum of stakeholders, fine tuning and further development of the process is geared towards benefiting the life sciences industry and the general consumer market.
The tools exist for companies to take the steps needed to reap the benefits of validation. Clearly, if you aren’t taking the necessary steps to compete, then your competitors are assuredly doing what they can to gain a market advantage. Understanding and early adoption of GAMP can increase a company’s competitive position, especially with the introduction/implementation of new technologies. By staying aware of technological innovations, companies are able to increase efficiency, minimize risks and reduce costs.


0 comments:
Post a Comment